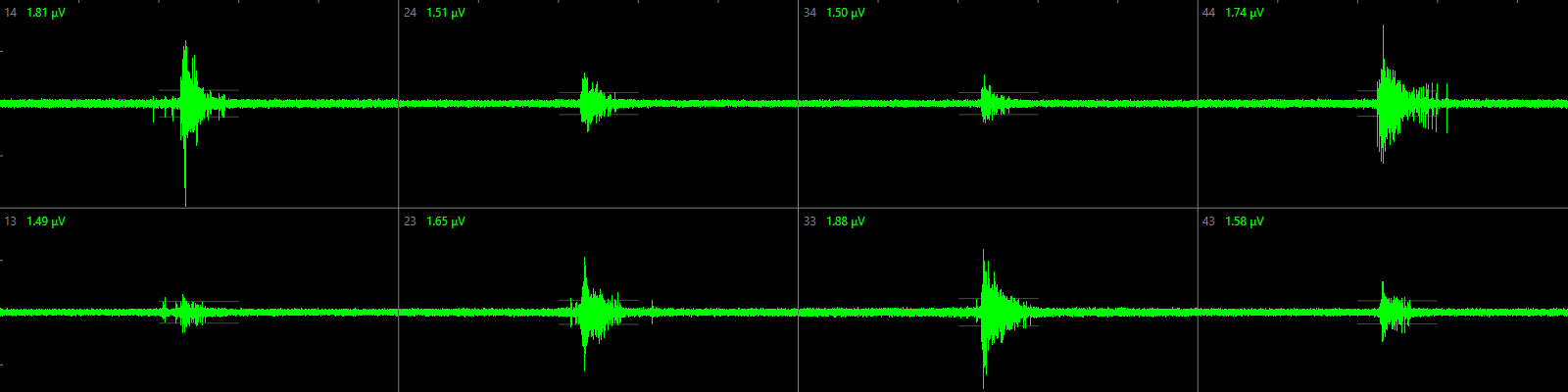
Micro-Electrode Array (MEA) recording of neuronal network activity
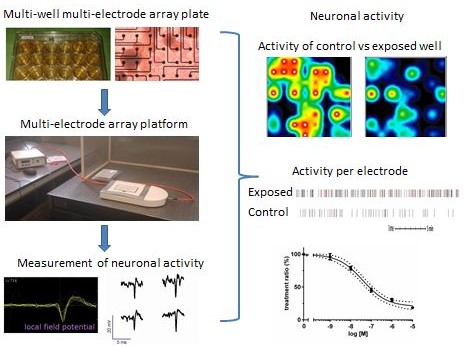
Chemical-induced changes in network function can be due to changes in electrical activity as well as to changes in sending or receiving intercellular signals. Micro-Electrode Arrays (MEAs) consist of a cell culture surface with an integrated array of 768 microelectrodes, allowing the recording of electrical activity in neuronal networks in vitro. We use a multi-well MEA system (Axion biosystems) to simultaneously record electrical activity at several locations in the neuronal cultures, thereby providing an integrated measure for chemical-induced effects on (the development of) neurotransmission within a neuronal network in vitro.
MEA approaches have only recently been introduced in neurotoxicological research, mostly for the investigation of acute disruption of network function (see e.g. McConnell et al., 2012). Recent investigations of the inter-laboratory variation in MEA recordings in rat cortical cultures has shown that neurotoxic responses can be measured reproducibly (Novellino et al., 2011).
We now apply this method for (sub)acute and chronic neurotoxicity, developmental neurotoxicity and neurodegeneration studies (see e.g. Dingemans et al., 2016; de Groot et al., 2013, 2014) and we recently used this approach even for food safety testing (Kasteel and Westerink, 2017; Nicolas et al., 2014) and hazard characterization of (illicit) drugs (Hondebrink et al., 2016, 2017). Moreover, it can also be applied for (collaborative) studies on cardiotoxicity or other studies using electroactive cell systems.
Spontaneous activity in rat neonatal cortical cultures or human iPSC-derived neuronal cultures can be measured in 12- or 48-wells MEA plates from 4-21 days in vitro using a MEA platform (left). Neuronal activity can be visualized as ‘activity maps’ per well as well as mean spike rate per electrode, resulting in concentration-effect curves that demonstrate the effect of neuroactive compounds (including (environmental) chemicals, drugs, food components) on neuronal activity.
Measurement of the intracellular calcium concentration
Proper calcium homeostasis is essential for neuronal function; prolonged increases in calcium will induce cell death, whereas transient increases in the intracellular calcium concentration can induce protein phosphorylation and gene expression. Importantly, also neurotransmitter secretion (exocytosis) is triggered by an increase in the intracellular calcium concentration (see e.g. Westerink, 2006). We therefore routinely measure changes in the intracellular calcium concentration using single cell fluorescent microscopy (see e.g., de Groot et al., 2014; Dingemans et al., 2010; Heusinkveld et al., 2010, 2016, 2017; Hondebrink et al., 2011; Meijer et al., 2014, 2015).
Cells are seeded in glass-bottom dishes and loaded with the high-affinity calcium-sensitive fluorescent dye Fura-2-AM prior to chemical exposure. Upon binding of calcium to intracellular Fura-2, the spectral properties of Fura-2 change resulting in a change in fluorescence. The change in fluorescence can thus be directly related to changes in the intracellular calcium concentration.
Noteworthy, single cell fluorescent microscopy outperforms plate reader-based high-throughput measurements of the intracellular calcium concentration, which are subject to numerous caveats, limitations and pitfalls (Westerink and Hondebrink, 2010; Heusinkveld and Westerink, 2011; Meijer et al., 2014).
Measurement of neurotransmitter secretion
The neurotoxicology research group has a long-standing expertise in measuring neurotransmitter secretion at the single cell level with single vesicle resolution using carbon fiber microelectrode amperometry (see e.g. Westerink et al., 2000; Westerink, 2004; Westerink and Ewing, 2008; Dingemans et al., 2009; Hondebrink et al., 2011; van Kempen et al., 2011).
A carbon fiber microelectrode is placed against the cell surface and used to instantaneously oxidize easily oxidizable neurotransmitters, such as dopamine. Following stimulation of the cell, vesicle fusion and neurotransmitter release (exocytosis) will occurs. The released neurotransmitters will be rapidly oxidized on the electrode surface resulting in a measurable electrochemical current, which is directly proportional to the amount of released neurotransmitter. Following recording, several specific parameters can be determined, including the frequency of exocytosis and the number of neurotransmitter molecules released per fusion event, to shed light on the characteristics of neurotransmitter release.
Measurement of receptor-mediated ion currents
Usually, ionotrophic neurotransmitter receptors require binding of the endogenous ligand (the neurotransmitter) to become activated. Therefore, these neurotransmitter receptors are also often referred to as ligand-gated ion channels. Activation and opening of these ligand-gated ion channels will result in an ion current across the cell membrane, which can be measured and using the voltage-clamp technique. These ion currents can be recorded in cultured neuronal cells as well as in Xenopus oocytes expressing the receptors of choice.
This test system can be used to assess the potential of chemicals to interfere with neurotransmitter receptor function. Following application of the endogenous ligand to test whether the cell expresses functional receptors, the cell can be superfused with test chemicals to test whether these can act as full agonist, i.e., are capable of activating the receptor in the absence of the endogenous ligand (see e.g., Antunes Fernandes et al., 2010). Additionally, chemicals may be capable of inhibiting (antagonist) or potentiating (partial agonist) the ligand-induced ion current (see e.g., Antunes Fernandes et al., 2010; Hendriks et al., 2010, 2012; Hondebrink et al., 2011, 2013, 2015; van Kleef et al., 2008).
Measurements of cell viability and oxidative stress
To exclude that effects are simply due to cytotoxicity, we routinely use a combined alamar Blue (aB) and Neutral Red (NR) assay to determine whether cell viability is affected by chemical exposure (e.g. Hendriks et al., 2012; Heusinkveld et al., 2010; Dingemans et al., 2010). With this method mitochondrial activity of the cells is recorded as a measure of cell viability with the aB assay, which is based on the ability of the cells to reduce (non-fluorescent) resazurin to (fluorescent) resorufin. Additionally, membrane integrity and lysosomal activity are subsequently determined as a measure of cell viability using the NR assay. NR is passively taken up in lysosomes, thereby providing an independent second measure for cell viability. Alternatively, we use a combined alamar Blue (aB) and CFDA assay. In that case, the presence of cytoplasmic enzymes in intact cells capable of de-esterification of non-fluorescent CFDA-AM to fluorescent CFDA is recorded as measure of cell viability. Alternatively, a MTT assay can be used to determine whether cell viability is affected by measuring the capacity of exposed and control cells to reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan (e.g. Dingemans et al., 2009; Hondebrink et al., 2009). Comparable with the Ab assay, the MTT assay is based on mitochondrial activity. These measurements of cell viability can be performed in multi-well plates, thereby enabling considerable throughput.

